Por Cristina Zita Díaz Pisano
Introducción
Introducción
La Enfermedad de Alzheimer consiste en una
afección neurodegenerativa
relacionada con el envejecimiento. Esta constituye, hoy en día, la causa más
frecuente de demencia en personas mayores de 65 años.
Clínicamente se manifiesta como un
síndrome, caracterizado por alteraciones en las funciones intelectuales
(memoria, lenguaje, atención, concentración, praxias, gnosias, funciones
visual-espaciales y capacidades ejecutivas), frecuentemente acompañado de alteraciones
psicológicas y de conducta, y que se traduce en la alteración en el
funcionamiento del enfermo con respecto al nivel que tenía previamente,
provocando situaciones de desadaptación social, laboral, familiar, etc., y la
dependencia para casi todas las actividades de la vida diaria. La EA tiene,
habitualmente, una instauración insidiosa y una evolución lentamente progresiva
que no permite detectar síntoma alguno en los primeros estadíos de la
enfermedad. Los primeros indicios clínicos que se pueden detectar son: olvido
de nombres, depresión, apatía anormal, etc.
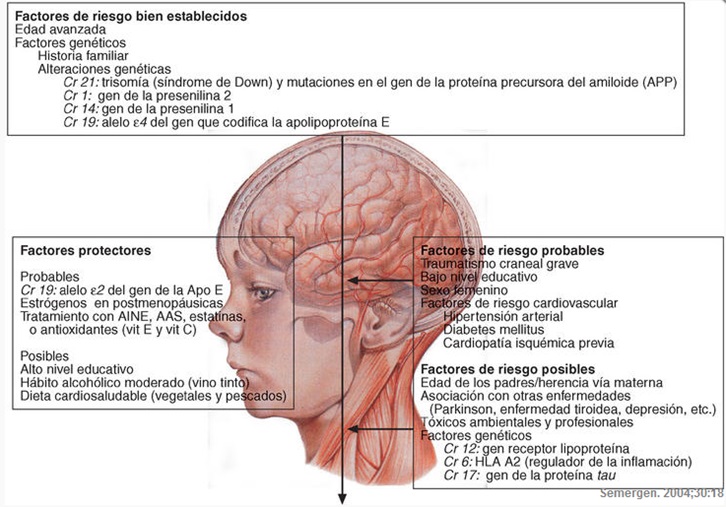
Factores
de riesgo
Existen ciertos factores de riesgo en relación con esta afección:
-Edad: Afecta a 1 de cada 9
personas +65 años y a 1 de cada 3 personas de +85 años. Aun no se ha esclarecido
completamente el por qué de este gran aumento con la edad.
-Historia familiar y genética:
el riesgo de padecer EA aumenta si algún miembro de tu familia. Esto se debe a
la herencia de genes. Tipos de genes implicados:
Ø Genes
de riesgo: Aumentan la probabilidad de desarrollar la enfermedad pero tenerlos
no garantiza padecerla. El de mayor influencia es una de las tres formas del
gen de la apolipoproteína E, el gen
APOE-e4, estimado como factor en un 20-25% de los casos.
Ø Genes
determinísticos: Se han observados variaciones en genes que causa la EA. Se
trata de tres proteínas: precursor de la proteína amiloide (APP), presenilina-1 (PS-1) y presenilina-2 (PS-2). Cuando son estos genes los
causantes de la enfermedad estamos tratando con Enfermedad de Alzheimer
autosómica dominante (ADAD), que supone un 2-5% de los casos de EA y se suele
presentar en el individuo antes de los 60 años.
-
Traumatismo craneoencefálico (TCE): TCE
lesiones moderadas y severas del cráneo/encéfalo aumentan el riesgo de desarrollar
EA en un 2 y 4.5 respectivamente. TCE moderadas implican pérdida de conciencia
o amnesia post-traumática que dura más de 30 min, si tal periodo es mayor de 24
horas se considera severa
-Padecimiento de enfermedades cardiovasculares:
Recientes estudios correlación la salud y buen funcionamiento de corazón y el
sistema cardiovascular con la disminución en el riesgo de padecer EA y otras
demencias. Así, aquellos hábitos que contribuyan a reducir el riesgo de padecer
enfermedades cardiovasculares también reducirán el riesgo de padecer demencia.
Son por ejemplo: actividad física moderada realizada regularmente, dieta
mediterránea equilibrada (baja en grasas saturadas y rica en verduras, frutas,
omega-3 ), peso adecuado, ausencia de infartos.
No obstante, por lo general, el desarrollo de esta
enfermedad se verá también afectado por factores ambientales, se pone
así de manifiesto el continuo debate ‘’nature .vs.nurture’’ que se presenta en la
patología actual.
Algunos científicos defienden la existencia de otros
factores que pueden influenciar en el EA. Estos son la educación y la inclusión
social del individuo. Aunque aún no se han presentado evidencias
científicas claras sobre la influencia de estos factores en el desarrollo de
EA, se piensa que el tener una Educación formal lo más amplia posible, así como
el mantenerse activo social y cognitivamente reduce el riesgo de padecer EA,
pues aumenta las conexiones entre las neuronas y permite al cerebro adaptarse
mejor a los posibles cambios, favorece la llamada reserva cognitiva.
Epidemiologia
Según datos proporcionados por la Organización Mundial de
la Salud (OMS), en el mundo entero, hay unos 47,5 millones de personas que padecen
demencia, y cada año se registran 7,7 millones de nuevos casos. La EA, que es la causa de demencia más común,
acapara entre un 60% y un 70% de los casos. De la totalidad de
enfermos, un poco más de la mitad (58%) viven en países de ingresos
bajos y medios.

Destacamos de la anterior gráfica que,
en general la prevalencia oscila entre el 4-6%. También es notable la
disminuida prevalencia de la enfermedad en China y el Oeste-Pacífico en
comparación con Norteamérica.
Se espera que durante los siguientes 20
años, si no se halla la cura de la enfermedad, los números de personas con
demencia incrementen en un 40% en Europa, un 63% en Norteamérica, un 77% en el
cono sur de Latinoamérica y un 89% en los países desarrollados de Asia
Pacífico.
De la misma forma, destaca la gran diferencia entre la mayor
afectación a mujeres que a hombres. Sobre este aspecto hablaremos más adelante.

Etiopatogenia
La etiología de la enfermedad es
desconocida.En función de la edad de aparición de los síntomas se clasifica en:
•
Enfermedad de Alzheimer de inicio precoz, si el comienzo es ante de los 65
años. En la
mayoría de las familias
con la forma presenil, hay un ligamiento a los marcadores del brazo largo del
cromosoma 14. La mutación en el cromosoma 14q parece originar un fenotipo más
grave que el causado por la mutación de la PPA
•
Enfermedad de Alzheimer de inicio tardío, si comienza después de los 65 años.
A su vez estas dos formas se clasifican
en dos subtipos:
•
Familiar, si hay historia familiar. El
gen de la enfermedad de Alzheimer se localiza en el brazo
largo del cromosoma 21.
Este hecho se relaciona con que los pacientes con trisomía 21 (síndrome de
Down) desarrollan con gran frecuencia el cuadro de la enfermedad y porque el
gen de la proteína precursora de amiloide (PPA) cerebral se localiza también en
ese cromosoma.
•
Esporádica, si no hay antecedentes familiares.
En la EA tienen gran importancia los
cambios degenerativos en el cerebro demostrables tanto por anatomía patológica
como por tomografía computarizada. Puesto que aún no se conoce la cura, su
tratamiento se basará sobre todo en tratar de mejorar la calidad de vida del
enfermo y retrasar el progreso de la enfermedad. Es importante también destacar
el rol que desempeña el apoyo del grupo familiar y la práctica de actividades
físicas e intelectuales estimulantes. Antes de entrar de lleno en el
funcionamiento, diagnóstico y tratamiento de esta compleja enfermedad
disfrutemos de esta sencilla y entretenida introducción a la afección.


Bibliografia
*La Demencia (Abril 2016)
publicado por la Organización Nacional de la Salud.
*Alzheimer disease: Epidemiology, Diagnostic
Criteria, Risk Factors and Biomarkers (Enero 2014) publicado por Biochem Pharmaco
http://www.ncbi.nlm.nih.gov/pmc/articles/PMC3992261/#R3
*Report 2014 and 2015 Alzheimer’s
Disease Facts and Figures statistical resource (U.S.A, 2014) publicado por Alzheimer’s
Association
*La Enfermedad del Alzheimer (Enero 2004) publicado por
Semergen-Medicina de familia http://www.elsevier.es/es-revista-semergen-medicina-familia-40-articulo-la-enfermedad-alzheimer-13056853
*Factores de riesgo para la EA, publicado por Alzheimer’s
Association http://www.alz.org/alzheimers_disease_causes_risk_factors.asp






